
«Pero sólo recientemente hemos podido profundizar en estas patologías gracias a los avances en tecnologías aplicadas, en técnicas de diagnóstico y en la disponibilidad de medicaciones eficaces», apunta la especialista del Hospital Universitario Fundación Jiménez Díaz (HUFJD).
Las enfermedades autoinmunes sistémicas (EAS) constituyen un grupo de procesos patológicos cuyo denominador común es la presencia de autoanticuerpos circulantes (proteínas del sistema inmune) capaces de causar equivocadamente diferentes deterioros en el cuerpo humano.
Las EAS más dañinas para el tejido intersticial pulmonar son las vasculitis sistémicas y las conectivopatías, como la artritis reumatoide (AR), esclerosis sistémica (ES), síndrome de Sjögren (SS), miositis inflamatorias idiopáticas (MII), enfermedad mixta del tejido conectivo (EMTC) o el lupus eritematoso sistémico (LES).
A la par, las enfermedades pulmonares intersticiales difusas (EPID), alrededor de 200 procesos heterogéneos, pueden converger, como vía final, en el desarrollo de fibrosis.
Unas EPID son de causa conocida y otras de causa desconocida; unas reflejan afectación exclusivamente en el pulmón y otras demuestran que el pulmón es un elemento más de deterioro en el contexto de alguna enfermedad sistémica.
En este sentido, la asociación de conectivopatía y EPID es más común en esclerodermia y miopatías inflamatorias idiopáticas.

Conferencia de la Dra. Olga Sánchez Pernaute, reumatóloga, sobre los efectos de las patologías autoinmunes sistémicas en el tejido conectivo pulmonar, enmarcada en el foro «Visionari@s: innovación y futuro en enfermedades respiratorias»
«A nivel etiopatológico, estas enfermedades lo que hacen es tener una pérdida de autotolerancia en la periferia: dermis, hipodermis, capas submucosas, membrana sinovial, paredes de los vasos sanguíneos, fluido seroso e intersticio pulmonar.
En esta área afectada por la inflamación se organiza una auténtica estructura linfoide terciaria (activación de la respuesta inmune humoral y celular), desarrollándose fenómenos de presentación antigénica y una expansión de clones linfocitarios autorreactivos (linfocitos T generados en el timo, órgano del sistema linfático).
Esto da lugar a que se genere una memoria y, por tanto, debemos saber que hay un riesgo aumentado de cronicidad y, consecuentemente, de que se desarrolle una fibrosis progresiva en el intersticio pulmonar, en este caso.
También, estas enfermedades autoinmunes van a cursar en brotes y remisiones, una realidad que enlaza con la intervención de la doctora Pérez Rojo, neumóloga, en su conferencia de visionarios: «El especialista debe saber cuándo y cómo tratar al paciente en cada momento evolutivo de la enfermedad».
Si tratamos al paciente con antifibróticos durante una exacerbación de la patología podremos, en cierto modo, impedir su progresión y minimizar el daño de la fibrosis.

Son pacientes complejos porque cada enfermedad del tejido conectivo tiene mecanismos fisiopatológicos diferentes y existen múltiples fenotipos dentro de un mismo diagnóstico clínico; es decir, es una población altamente heterogénea.
De hecho, la presentación clínica de las EPI-EAS engloba tanto a pacientes asintomáticos como a otros con disnea grave.
Pero al final, la enfermedad que progresa negativamente compartirá mecanismos lesionales.
L@s especialistas, especialmente en neumología, están muy acostumbrad@s a ver estas estructuras al diagnosticar y tratar estas patologías pulmonares, puesto que, seguramente, son comunes a todas ellas.
Tanto es así que desde hace algunos años la cultura médico-científica ha establecido que la fórmula más eficaz para atender las intersticiopatías es el análisis clínico multidisciplinar.
Y cabe destacar que, una vez cribados y sobre todo descartados los agentes etiológicos más frecuentes, subyace un proceso autoinmune en un número no desdeñable de pacientes; bien en forma de patología del tejido conectivo o bien en lo que más recientemente se ha denominado IPAF (neumonía intersticial idiopática con características autoinmunes, entidad diferenciada entre las EPID).
Con estas estrategias, debemos conseguir un diagnóstico lo más detallado y preciso posible, tratando de establecer los mecanismos fisiopatológicos causales y seleccionando a los pacientes meticulosamente para encuadrarlos en las líneas de tratamiento disponibles.
También, en alguna medida, debemos lograr el mejor pronóstico o prever el riesgo de progresión.
A este respecto, cuando se introdujo el término de IPAF nos ayudó mucho, aún más a la neumología, realizar una especie de lista de requisitos (checklist de ítems) para valorar en l@s pacientes las posibles manifestaciones extrarespiratorias asociadas a su enfermedad.
Con estos principios, derivamos a los pacientes a la consulta monográfica en función de la existencia de algunas de estas alteraciones, ya sea en los dominios radiográfico, clínico o serológico.

En un nivel radiográfico, básicamente, se parte de patrones no NIU o de patrones NIU (neumonía intersticial usual) que tengan algunas características atípicas, como la presencia de signos extraparenquimatosos.

Mientras que las manifestaciones clínicas, como se observa en la fotografía, aparecen muy asociadas a enfermedades del tejido conectivo: artritis, Raynaud, esclerodactilia, úlceras digitales, telangiectacias, exantema fijo en las superficies extensoras, lesiones hiperqueratosas en manos o edema digital.
Además, en el campo serológico buscaremos la existencia de algún autoanticuerpo (auto-AC) típico de estas enfermedades en el plasma de los pacientes, como FR, ACPA, dsDNA, Sm, ACL, Ro60, La, U1RNP, MSA, ATA, ACA, etc.
Retos en EAS-EPID: anticipación a la fibrosis pulmonar a través del grandísimo valor de los autoanticuerpos
En primer lugar, los pacientes que no tienen dominio clínico debido a que estas manifestaciones, a veces, pueden ser sutiles, estar totalmente ausentes o haberse enmascardo previamente debido a un tratamiento farmacológico a base de corticoides.
En estos pacientes, que llamamos monoorgánicos o con afectación pulmonar dominante, encontraremos con cierta frecuencia autoanticuerpos, que son de gran utilidad porque tenemos la suerte de que se relacionan con el fenotipo clínico y, además, predicen lo que le va a ocurrir en la envolución de su enfermedad.
Incluso en pacientes que cumplirán criterios de una conectivopatía no es raro que estas manifestaciones debuten como formas aisladas a nivel respiratorio; y esto es todavía más frecuente en pacientes con neumonía intersticial idiopática con características autoinmunes (IPAF).

Las situaciones que nos podemos encontrar en un paciente monoorgánico serían las que se observan en la caja de la izquierda de la ilustración, es decir, portadores de autoanticuerpos específicamente vinculados a la intersticiopatía.
La segunda caja refleja los autoanticuerpos especialmente asociados a enfermedad del tejido conectivo, pero no tanto a la enfermedad intersticial.
En estos casos tendremos que indagar sobre otros factores que se puedan relacionar con lo que ocurre en los pulmones, como pueden ser microaspiraciones, tóxicos, ambientales, fármacos, etcétera.
Y en la tercera caja de la ilustración superior, a la derecha, situamos los anticuerpos de significado incierto porque no son tan específicos; con lo cual, desconocemos, como decía la doctora Pérez Rojo, qué tiene el paciente debajo de esa interciopatía acompañada de un anticuerpo.
Caso de los anticuerpos anti-sintetasa
Estos anticuerpos son muy interesantes porque definen un fenotipo clínico característico de pacientes que tienen intersticiopatía (EPID), miositis, artritis, fenómeno de Raynaud, fiebre, manos de mecánico y otras peculiaridades.
Aún así, el paciente no suele tener todas estas manifestaciones, sino que vemos muchas formas incompletas:
Algunos no encajan en los criterios clínicos de una conectivopatía y otros pueden ser falsamente diagnosticados, por ejemplo, de una artritis porque predomina esta manifestación o de un síndrome Sjögren.

En verdad, observamos un gran solapamiento, por ejemplo, entre los criterios propuestos para IPAF y los criterios propuestos para el síndrome antisintetasa (miopatía inflamatoria idiopática, afectación intersticial pulmonar, artritis no erosiva y manos de mecánico).
En nuestra cohorte retrospectiva AIP HUFJD se observa que de los pacientes portadores de estos anticuerpos algunos encajaban en la clasificación de IPAF mientras que otros lo hacen en el listado de miopatías inflamatorias.
Esto hizo proponer al Grupo Nerea de la Comunidad de Madrid, al cual pertenezco, y a otros grupos multidisciplinares seguramente les habrá pasado lo mismo, que deberíamos diferenciar a los IPAF que tienen estos anticuerpos del resto de IPAF, ya que fisiopatológicamente, casi con seguridad, no tienen nada que ver entre sí.
Tanto es así que no tendría mucho sentido, por ejemplo, hacer un ensayo clínico mezclando estas dos poblaciones o cohorte de pacientes.
Otra peculiaridad es que analizamos a pacientes con anticuerpos muy específicos, nuevamente los sintetasa, que tienen los patrones NIU: lineal-reticular, nodular, de vídeo deslustrado, quístico y de condensanción.
Y si un paciente debuta de forma monoorgánica y tiene un patrón NIU inicialmente no vamos a pensar que estamos ante una enfermedad autoinmune. ¿Y cómo se actúa cuando vemos un anticuerpo tan específico?

En las cohortes de pacientes publicadas sobre el tema (por ejemplo, en esta ilustración con anti-PL-12, que es un anticuerpo antisintetasa) se ha comprobado, efectivamente, que una gran mayoría de ellos y ellas debutaban de forma aislada a nivel respiratorio.
Complementariamente, la mayoría de los patrones eran considerados NIU o fundamentalmente NIU.
Es decir, si el paciente tiene un anticuerpo antisintetasa es indiferente que cumpla o no los criterios de IPAF, ya que posiblemente tiene una enfermedad autoinmune de base.
Esta propuesta, por supuesto, todavía no está validada y necesitaríamos más estudios rotundos, sobre todo confirmatorios a nivel histopatológico.

Una rutina diagnóstica que empleamos nosotr@s en la consultas sería el cribado de pacientes con estudios capilaroscópicos… ¿Por qué?
En los ítems clínicos del cuadro ‘EPID posiblemente autoinmune (NO IPAF)’ -ilustración en zona superior- se observa que todas las variables, salvo la artritis, se asocian a vasculopatía digital en el dominio clínico de los pacientes con IPAF.
Realmente, esta vasculopatía es un rasgo muy típico de pacientes con enfermedad del tejido conectivo, sobre todo en el espectro de la esclerosis sistémica, pero también del síndrome antisintetasa, de la dermatomiositis y otras menos caracterizadas.
Para las médicas y los médicos efectuar una capilaroscopia en un paciente monoorgánico o con una enfermedad de significado incierto nos puede proporcionar información de altísimo valor.
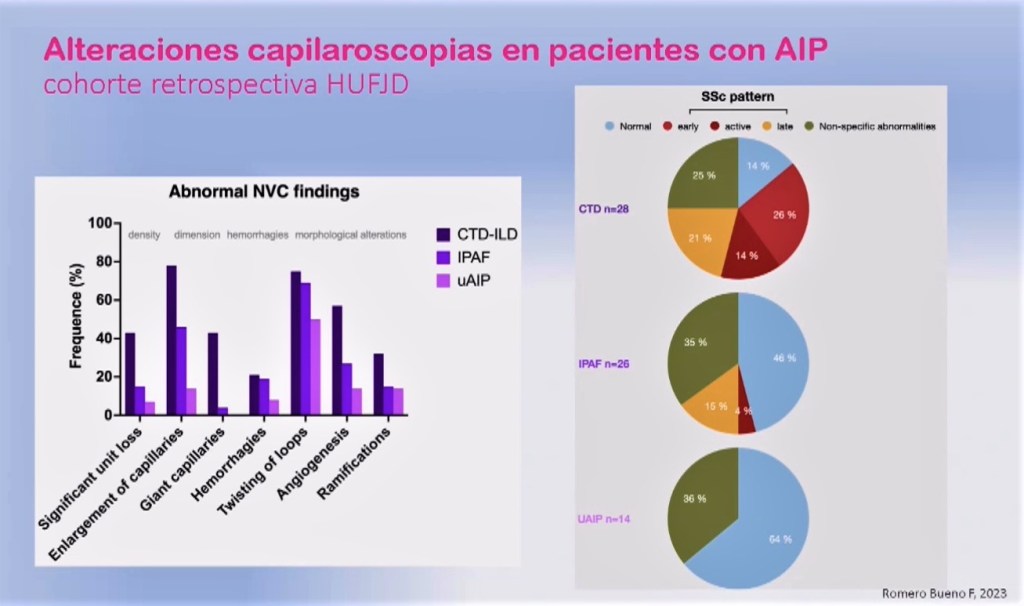
Como ejemplo, valga nuestra cohorte retrospectiva HUFJD, donde analizamos cuáles eran los resultados de la capilaroscopia en pacientes con enfermedad intersticial: tanto pacientes con IPAF, es decir, que no cumplen criterios clasificatorios de enfermedad del tejido conectivo, como incluso aquellos que llamamos no clasificables porque ni siquiera cumplen criterios IPAF, algunos sí que demuestran alteraciones características en la capilaroscopia que les permiten clasificarlos prácticamente como una conectivopatía.
Es más, el síntoma guía característico, que es el fenómeno de Raynaud, no se observa en todas las ocasiones. Son pacientes que tienen otras lesiones digitales o ni siquiera lesiones; y por dentro, cuando los miramos, sí tienen estas alteraciones.
El valor de las pruebas que buscan la precisión diagnóstica absoluta
En el cribado de los pacientes con una intersticiopatía, sobre todo si es atípica o no tiene un diagnóstico claro, será decisivo realizar la prueba diagnóstica de anticuerpos antinucleares en sangre.
Eso sí, es importante hacer un cribado bastante completo; no nos vale con una técnica de ‘screening‘ como puede ser ELISA (deteccción en sangre de biomarcadores), que es la que habitualmente tienen todos los servicios de inmunología.
Hay que llevar a cabo, también, una inmunofluorescencia, que debe guiar la búsqueda de antígenos específicos relacionados con la enfermedad intersticial.
Y como nos enfrenamos a una enfermedad rara, donde los pacientes no abundan, vale la pena, y mucho, hacer un abordaje individualizado, tener una conexión directa con inmunología, explicarles la sospecha clínica e intentar cribar el anticuerpo.
Ahora bien, nos topamos con un problema, ya que se han puesto en marcha ensayos comerciales cuya validación no es muy buena.
No conocemos bien su sensibilidad y especificidad: podemos tener falsos positivos o múltiples positividades, lo cual suele ser habitualmente un signo de baja especificidad, o podemos tener anticuerpos no detectables.
Por tanto, ante una sospecha clínica alta sin un anticuerpo que plasmar en el informe médico, debemos mantener la sospecha y tratar de cribar al paciente hasta despejar cualquier duda razonable.
Dra. Olga Sánchez Pernaute, reumatóloga de la Fundación Jiménez Díaz y del Grupo Nerea de la Comunidad de Madrid

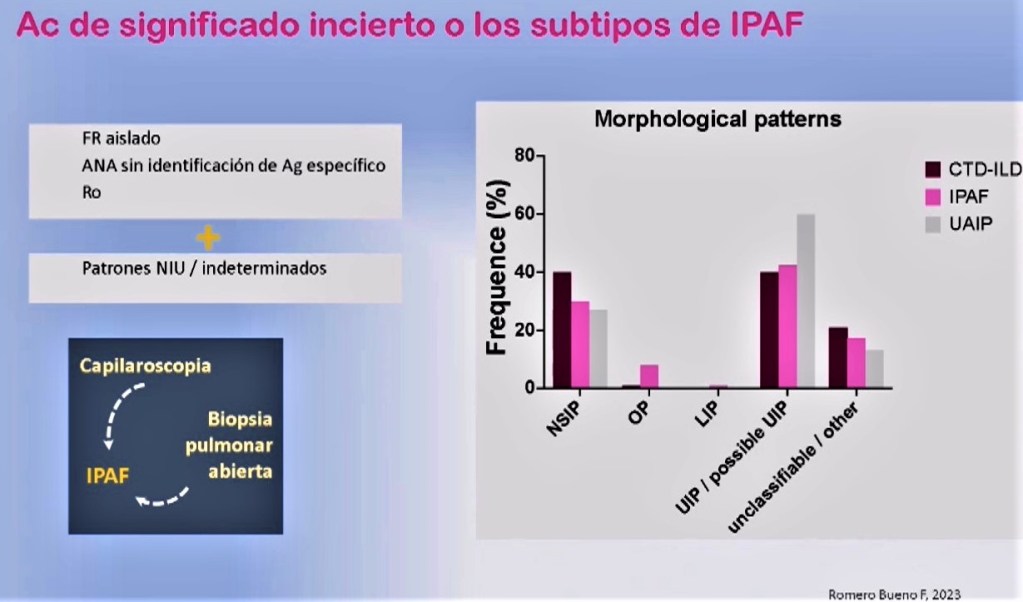
¿Y qué sucede con los anticuerpos de significado incierto o subtipos de IPAF, e incluso con los patrones NIU que ni siquiera cumplen este criterio de IPAF?
Volviendo a nuestra cohorte HUFJD, si observamos el patrón radiográfico predominante, que curiosamente era NIU o posible NIU, no está tan claro que debamos cribar solamente las NINE (enfermedades intersticiales no específicas, inflamatorias de base): existen neumonías intersticiales usuales (NIU) que tienen un sustrato autoinmune que también deben ser cribadas.
¿Y cómo diagnosticarlas?… Habrá veces que ni siquiera vayamos a cumplir los criterios de IPAF, por lo que nuevamente, como hemos visto antes, recurriremos a la capilaroscopia. En estos casos, también creo que debemos plantear la oportunidad de realizar una biopsia pulmonar abierta.

En conclusión, no tenemos fenotipos claros en las patologías EAS-EPID, sino que existe un gran solapamiento y muchas formas de afectación pulmonar dominante, por lo que se necesitan los datos de la práctica clínica para progresar en el diagnóstico, el tratamiento y el pronóstico de estas enfermedades raras que pueden acabar en fibrosis.
Este videoblog se ha elaborado a partir de la intervención de la Dra. Olga Sánchez Pernaute en la mesa de debate “Fibrosis Pulmonar Progresiva” del programa científico “Visionarios, Innovación y Futuro en Enfermedades Respiratorias”; un encuentro profesional que se desarrolló el 6 de octubre de 2023 en el salón de actos del Hospital Universitario de La Princesa de Madrid.




Debe estar conectado para enviar un comentario.